Alain Pralong1,3 Renaud Jacquemart2,3 Katrina Cordovado2,3
- Pharma Consulting ENABLE GmbH
- Omnium Global Consulting Inc.
- ENABLE Biotech AG
Bringing a Cell or Gene Therapy from the lab to its First-in-Human (FIH) trial is an exciting milestone—but getting there can feel like navigating a regulatory maze. The promise of these therapies to cure previously untreatable diseases is immense, but the journey is filled with potential pitfalls. Understanding the key regulatory requirements early can make the difference between a smooth transition to the clinic and costly delays. So, how can CGT companies chart a clear path through this complex process?
The TYPICAL REGULATORY PROCESS FOR FIH TRIALS
Conducting Trials in Europe
Companies must submit a Clinical Trial Authorization (CTA) to the EMA for centralized approval. The CTA includes detailed information on preclinical data, manufacturing processes, and clinical trial protocols.1 For companies conducting trials in Switzerland, for example, the regulatory journey involves submitting a CTA to SwissMedic, the Swiss Agency for Therapeutic Products, and concurrently obtaining independent ethics approval from the trial site’s committee. During the pre-clinical stage, companies are recommended to engage with SwissMedic for scientific advice on study design and regulatory expectations2. Conducting trials in Switzerland can offer several advantages, including faster approval times compared to the EMA and mutual recognition agreements with the EU & US.
When conducting a trial in other European countries, the national regulatory agencies would be activated as rapporteurs for a centralized EMA submission. Each of these agencies will then be assessed according to the EMA’s guidelines for member states.
The centralized review process by the EMA for CGTs ensures that FIH trials are scientifically sound and ethically justified, while supporting manufacturers and even accelerating the process for certain products4. Throughout all stages, ensuring compliance with Good Manufacturing Practice (GMP) standards is crucial. This involves stringent controls over sourcing, production, and quality assurance processes5. Regular inspections of CDMOs by local regulatory authorities help maintain GMP certification, guaranteeing high-quality production standards6.
Conducting Trials in the US
In the United States, the regulatory pathway for FIH trials begins with the submission of an Investigational New Drug (IND) application to the FDA. This application must include comprehensive preclinical data, details of the manufacturing process, and clinical trial protocols. The FDA reviews the IND to ensure that the proposed trial can proceed safely, focusing on aspects such as the starting dose, dose escalation plans, and safety monitoring strategies7.
The FDA offers pre-IND meetings to provide guidance and feedback on trial design and development plans, as well as expedited review processes for therapies that show promise in treating serious or life-threatening conditions.
PARTNERING WITH A CDMO: YOUR KEY TO REGULATORY SUCCESS IN FIRST-IN-HUMAN TRIALS
There’s no doubt: navigating the regulatory landscape for FIH trials in CGTs can be daunting. CDMOs play a critical role in de-risking and accelerating the FIH)trial process by ensuring that regulatory submissions meet the highest standards. Their expertise in CMC documentation, GMP compliance, and regulatory interactions makes them invaluable partners for biotech companies entering clinical trials.
The journey to FIH can be acclerated with selection of the right CDMO focused on:
- Tailored CMC Strategies: Customization of the CMC section to align with the specific requirements of various regulatory bodies, ensuring a smooth approval process.
- Regulatory Submission Support: Expert guidance on navigating the regulatory landscape of various jurisdictions, from initial scientific advice to final application submissions, Supporting biotech companies in preparing for pre-IND (FDA) or scientific advice meetings (EMA/SwissMedic), ensuring alignment with regulatory expectations.
- Clinical Trial Management: Connections to hospitals, clinical investigators, source materials, and a network of regulatory experts, accelerating initiation of trials and ensuring adherence to GCP guidelines and regulatory standards.
- GMP Manufacturing & Compliance Support: Updated facility qualifications, as well as support with preparing batch records, stability data, and validation reports to ensure compliance with GMP guidelines from EMA, FDA, and SwissMedic.
- Global Expansion Support: Advising biotech companies on the best jurisdictions for trial initiation and helping US companies navigate EU regulatory pathways.
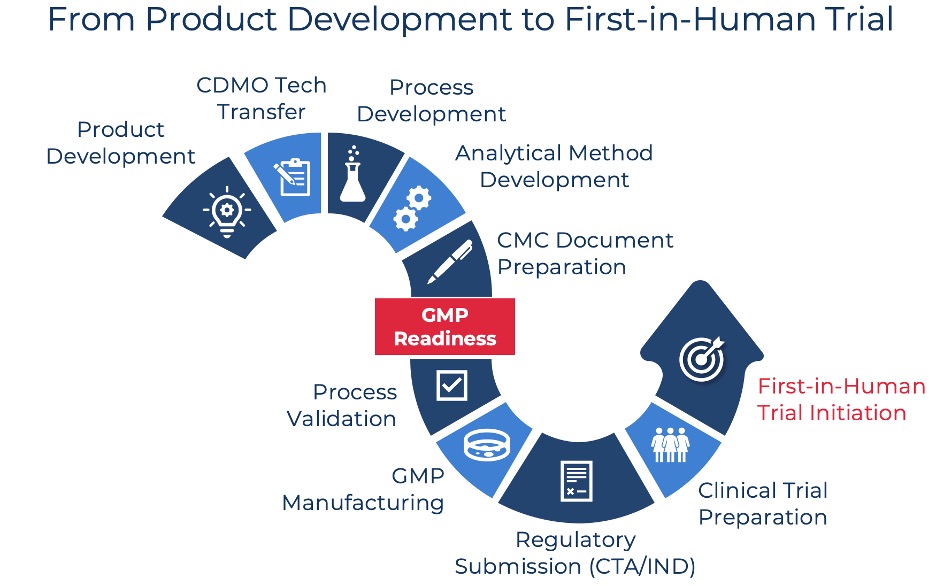
Navigating the complexities of FIH cell therapy trials requires a deep understanding of the regulatory environment, strategic planning for clinical trials, and compliance with GMP standards. ENABLE Biotech is equipped to manufacture for companies conducting trials in both EMA and FDA regulated areas. Our facilities and processes are designed to comply with ICH guidelines, providing a solid foundation for meeting diverse regulatory requirements. We leverage our CMC expertise to assist companies in submitting their CTAs in the jurisdiction of their choice, and can also support US companies looking to enter European markets with initiating their trials in Switzerland. Switzerland’s strategic location and robust regulatory framework offer a favorable environment for CGT development and commercialization.
Choosing the right CDMO partner can mean the difference between a smooth regulatory submission and costly delays. ENABLE Biotech provides end-to-end regulatory support, from CMC strategy and GMP manufacturing to guiding companies through EMA & FDA requirements. Our expertise ensures that clients avoid regulatory pitfalls, accelerate their path to FIH trials, and bring transformative therapies to patients faster. If you are currently preparing to bring your ATMP to patients for the first time, reach out to us to discuss how our CDMO services can streamline your regulatory journey!
References
1. European Medicines Agency (EMA). Guidelines on Advanced Therapy Medicinal Products (ATMPs). Available at: [EMA website] (https://www.ema.europa.eu/en/human-regulatory/overview/advanced-therapy-medicinal-products-overview)
2. Swissmedic. Clinical Trial Application. Available at: [Swissmedic CTA] (https://www.swissmedic.ch/swissmedic/en/home/humanarzneimittel/clinical-trials.html)
3. EMA. Committee for Advanced Therapies (CAT). Available at: [EMA CAT] (https://www.ema.europa.eu/en/committees/advanced-therapies)
4. European Commission. Good Manufacturing Practice (GMP) Guidelines. Available at: [GMP guidelines] (https://ec.europa.eu/health/documents/eudralex/vol-4_en)
5. EMA Guideline on Quality, Non-Clinical and Clinical Requirements for Investigational Advanced Therapy Medicinal Products in Clinical Trials. European Medicines Agency. https://www.ema.europa.eu/en/guideline-quality-non-clinical-clinical-requirements-investigational-advanced-therapy-medicinal-products-clinical-trials-scientific-guideline
6. FDA Considerations for the Design of Early-Phase Clinical Trials of Cellular and Gene Therapy Products. U.S. Food and Drug Administration. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-design-early-phase-clinical-trials-cellular-and-gene-therapy-products